











There are many challenges involved when implementing a QMS system that will comply with ISO13485:2016. Here are top 10 requirements you should focus on early on, so that it will become easier and quicker to set up the systems and achieve the necessary compliance.

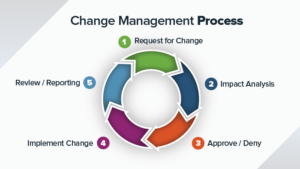
Key Requirements for Document Change Control

The documents and records used in the QMS are to be controlled to meet the requirements for ISO13485:2016.
You will need to create procedures to manage the Document Control and Change Control. For example :
- SOP0001: Good Documentation and Record Keeping Procedure.
- SOP0002: Document Control and Change Control Procedure.
Key Requirements for Document Change Control ISO13485 Section 4
When making changes for a document, following areas are to be focused.
- The Review and approval of the documents are done to ensure adequacy prior to use. Make sure all the new documents or changes to existing documents are review and approved prior to usage of the documents.
- Ensure that the current revision and changes to the current documents are identified.
- After the review and approval is done, ensure that the current version of the documents are available at the point of use. The
previous versions shall have been removed from the point of use. - Ensure that the documents remain legible and readily identifiable.
- Ensure that the documents of external origin are identified, and the distribution is controlled.

Additional Requirements for Document Change Control ISO13485 Section 4

- Prevent deterioration and loss of documents.
- Prevent unintended use of the obsolete documents and apply suitable identification to them.
- When changes are made to the documents, such changes are to be reviewed and approved by the original approving function or another designated function that has similar background.
- Have a documented procedure that defines the retention period for the obsolete documents.
How can Qualcy eQMS help for the Document Change Control?
- The Qualcy eQMS uses virtual cabinets for segregation of the documents by the approval and current status.
- Only the latest versions of the approved and effective documents are available in the Released cabinet. The previous versions are automatically stored in the Archived cabinet.
- Any documents that is being currently edited or revised is stored in Draft Cabinet.
- The document changes are reviewed by the original approving function or another designated function, through use of Approval Matrix in the Qualcy eQMS.
- Esign and audit trails maintained for document change control.

More Options for Document Change Control in Qualcy eQMS

- The records created in the Qualcy eQMS are assigned with unique ID numbers. The records are segregated based on the status.
- The change control provides option for impact assessment and linked to Risk Management. There is option of task assignment for verification and validation management.
- The document changes can trigger auto notification to the impacted employees through the integrated training plans in the Qualcy eQMS.
- The Qualcy eQMS provides options for fillable form that can be customized and used for various record keeping options.

Requirements for Management Responsibility: ISO13485 Section 5
- Management Commitment : The management team at your company shall demonstrate leadership and commitment with respect to quality management system to comply with ISO 13485:2016 requirements. How do you do this?
- Involve Management Team by taking accountability for the development and effectiveness of the Quality Management System.
- Ensure that the Quality Policy and Quality Objectives are established and in alignment with the context and direction of the organization.
- Ensure that the quality management system requirements are included into organization’s business processes. Create documented procedure for Management Review.
- Promote the process approach and implement risk management process.

Additional Requirements for Management Responsibility: ISO13485 Section 5

- Ensure the resources needed for the quality management system are available.
- Communicate the importance of effective quality management and of conforming to the quality management system requirements.
- Ensure that the quality management system achieves its intended results through management review.
- Engage and direct persons to contribute to the effectiveness of quality management system.
- Promote improvement through relevant management roles to demonstrate their leadership as it applies to their areas of responsibility
How can Qualcy eQMS help for ensuring Management Responsibility: ISO13485 Section 5 ?
- The Qualcy eQMS provides metrics and graphs that are used for Management Review meeting.
- The metrics include:
I. CAPA aging reports,
II. Trends for CAPAs by process areas, Root causes, departments and functional areas.
III. Trend for customer complaints by product lines, process areas and others.
IV. Trends for Internal Audit observations by process areas.
V. Trends for training completion be departments and functions,
VI. On-time performance of Calibration and PM events, trends for out of tolerance records.
VII. Status of supplier qualification records, performance of the Approved supplier list and others.


Requirements for Resource Management: ISO13485 Section 6

- You will need to determine the resource requirement and provide resources for the implementation, maintenance and continual improvement of the QMS.
- The management team has to identify the resources needed for all the functions of the organization to meet the quality objectives.
- The resources include physical infrastructure, buildings, equipment, computer hardware and software.
- The resources will be acquired and maintained under suitable controls to produce outputs that meet the customer requirements and regulatory requirements.
You will need to create documented procedures to control the Resource Management. For example :
> SOP0003: Procedure for Employee Qualification and Training Management.
> SOP0002: Procedure for control of Infrastructure and Equipment Management
Requirements for Human Resource Management: ISO13485 Section 6
- Your company shall provide human resources for the effective
implementation of the QMS and for the operation and control of its processes.How do you do this?- Determine necessary competence for the employees performing work that may impact the product quality. Create job descriptions and employee qualification requirements.
- Provide training and take actions to maintain the competency.
- Evaluate the effectiveness of the trainings done.
- Ensure that employees are aware of their role in the QMS and how they can impact the objectives of the QMS.
- Maintain records for education, training, skills and experience
- Determine necessary competence for the employees performing work that may impact the product quality. Create job descriptions and employee qualification requirements.

How can Qualcy eQMS help for the Human Resource Management: ISO13485 Section 6

- The Qualcy eQMS Training management module provides option for managing the employee competence, qualification, training and awareness.
- There is option for managing the Training matrix by job roles and training plans by employee id.
- The training plans are created for the individual users, the list of SOPs are added to the training plans.
- The effectiveness of the training plans is evaluated by the managers on the auto schedule.
How can Qualcy eQMS help for the Physical Resource Management: ISO13485 Section 6
Physical Resource Management:
- The physical resources, utilities and other infrastructures are assigned with unique asset ID numbers. The status, location and owner of assets are managed in the Qualcy Equipment module.
- The records for Calibration, PM, Work Orders and Validation Reports are maintained.
- The physical resources are acquired based on their suitability and qualifications to meet the process requirements.
- The physical resources are evaluated periodically to make sure they are capable of meeting the process requirements. This can be done by calibration and preventative maintenance of the resources.


Requirements for Design and Development: ISO13485 Section 7.3

- The requirements under this section are used as guidance for design, development, and release of new products and changes in the design of existing products.
- Your company shall establish, implement and maintain documented procedures for the design and development process that will ensure proper release of the products and services.
- Your company will determine the stages and the controls needed for the design and for the development process.
Requirements for Design and Development: ISO13485 Section 7.3
During the design and development process, you will need to consider the following:
- The nature, duration and complexity of the design and development activities.
- The required number of process stages and reviews.
- The required verification and validation activities.
- The required roles and responsibilities.
- The internal and external resource needs.
- Define the controls needed when multiple persons
interface. - Need for involvement of customers and users.

Additional Requirements for Design and Development: ISO13485 Section 7.3

Requirements for design controls include:
8.Design input requirements
▪ Functional, performance, usability and safety requirements, regulatory requirements, outputs from risk management and more.
9.Design output requirements
▪ Meet the input requirements for design, develop specs for purchasing, production and service provisions.
8.Design Review requirements
a)Evaluate the ability of the results of design and development to meet the input requirements;
b) Identify and propose necessary actions
c) Keep the records for design review
Additional Requirements for Design and Development: ISO13485 Section 7.3
Requirements for design controls include:
9. Design verification requirements
▪ The design and development outputs have met the design and development input requirements.
▪ The documented verification plans shall include methods, acceptance criteria and, as appropriate, statistical techniques with rationale for sample size
8. Design Validation requirements
▪ The resulting product is capable of meeting the requirements for the specified application or intended use.
▪ The documented validation plans that include methods, acceptance criteria and, as appropriate, statistical techniques with rationale for sample size
How can Qualcy eQMS help for the Design and Development: ISO13485 Section 7.3
- The Qualcy eQMS provides controls required for design and development of the new products and processes.
▪ The design requirements and process requirements are developed using fillable templates for creation of design input and design output documents. The design trace matrix provides complete traceability to design output results, verification and validation results.
▪ The design requirements are linked to the hazards associated with the medical device Risk management.
▪ The Risk Management includes Risk analysis, Risk control, Risk Verification and Validation.
▪ The risk trace matrix is integrated with design input requirements. - The design History folder creates table of content folder that combines all inputs and out puts of the design history file.

Requirements for Purchasing Controls: ISO13485 Section 7.4
Purchasing process:
- The process for purchasing of product and services are to be controlled.
- You will need documented procedures for supplier qualification and performance monitoring.
- You will need documented procedures for receiving of products and services to ensure that these products, and services are aligned with internal requirements.

Requirements for Purchasing Controls: ISO13485 Section 7.4

The purchasing requirements shall:
▪ Determine and apply criteria for the evaluation, selection, monitoring of the performance and reevaluation of suppliers, based on their ability to provide processes or products and services to comply with the requirements.
▪ Retain records of these activities and take any necessary actions arising from the evaluations. The Organization will continue to monitor and re-evaluate the suppliers in periodic intervals.
▪ Apply classification of the purchased material and services into criticality groups depending upon their impact to the final product quality and performance.
▪ Apply risk-based approach for the evaluation, selection, monitoring of the performance and reevaluation of external providers, based on their ability to provide processes or products and services to comply with the requirements.
Additional Requirements for Purchasing Controls: ISO13485 Section 7.4
Purchasing Information:
▪ Create specifications for the products and services.
▪ Acceptance criteria for product and services including support services and facilities as specified.
▪ Qualification requirements for supplier employees.
▪ Quality system requirements for the supplier.
▪ Written agreement that the supplier notify the organization of changes in the purchased product prior to implementation of any changes that affect.
▪ Communicate such requirements to the supplier and get approval from the suppliers.
▪ Maintain documented procedures for managing the above requirements.

Requirements for Purchasing Controls: ISO13485 Section 7.4

You are required to take actions when there is non-conformance observed for the purchased product and service.
▪ The actions shall include appropriate investigation and corrective actions. These actions shall be will be handled using documented procedures. For example:
▪ SOP0007: Out of Specification Policy and Procedure,
▪ SOP0011: Corrective and Preventative Action Procedure.
Requirements for Purchasing Controls: ISO13485 Section 7.4
Verification of purchased product:
▪ The process for inspection and verification of purchased product has to be documented in a procedure, for example: SOP0012: Incoming Materials Receiving and
Inspection policy.
▪ The inspection and verification shall be done as per the approved specifications.
▪ Maintain the records for the results of verification of purchased product. (For example: QF0002: Receiving Inspection Log)
▪ When change notifications are received from the suppliers, you will need to evaluate the impact on the quality and performance of the final product.

How can Qualcy eQMS help in Managing Purchasing Controls: ISO13485 Section 7.4?

The Qualcy eQMS Supplier Management module provides option for managing the evaluation and selection of the suppliers.
▪ Maintain approved supplier list and supplier profile.
▪ The ongoing evaluation of the suppliers are implemented through auto schedule.
▪ The use of fillable templates enables easy scoring and analysis of supplier quality system assessments.
▪ You can share the platform with suppliers for interaction and collection of inputs directly from the suppliers.

Requirements for Complaint Handling: ISO13485 Section 8.2
You will need to create a documented procedure for handling of the customer complaints, for example: (SOP0061: Handling of the customer complaints).
The procedure for complaint handling includes requirements and responsibilities for
▪ Receiving and recording information about the complaint.
▪ Evaluating the validity of the complaint.
▪ Evaluating the requirements for notifying the regulatory authorities
▪ Investigating complaints.
▪ Evaluating if related products have been impacted.
▪ Risk assessment for corrections and corrective actions.

Additional Requirements for Complaint Handling: ISO13485 Section 8.2

When there is no investigation is done, then justification for not doing the investigation are to be documented.
▪ When correction or corrective actions are implemented, such actions are to be documented in the Complaint reports. If CAPA records are opened, such records are to be referenced in the Complaint records.
▪ When the result of investigation confirms that the 3rd party service providers , e.g. shipping and handling companies, have contributed to the complaint, you
will need to notify the 3rd parties involved and the records for notification are maintained.
▪ The records for complaint handling process are to be documented in a form, e.g QF0071 (Customer complaint Form).
Requirements for Complaint Handling: ISO13485 Section 8.2 in Qualcy eQMS
The Qualcy eQMS complaint management module provides option for recording, evaluating, and investigating complaints.
▪ The complaint record include:
▪ Source of the complaint.
▪ Evaluate complaints- valid vs. invalid.
▪ Evaluate whether a complaint warrants reporting to regulatory bodies under FDA 21 CFR Part 803 and/or ISO 13485:2016 section 8.2.3 requirements.
▪ investigation required (yes/no).
▪ Records for the investigation completed.
▪ Reference to the CAPA record.


Requirements for Internal Audit: ISO13485 Section 8.4

- You will need to create a documented procedure for conducting internal audits in scheduled intervals, i.e. SOP0012 (Internal Audit Procedure),
- The purpose of the internal audit is to evaluate the effectiveness of the QMS against the requirements for ISO13485 and the requirements for the internal business process.
- The Internal Audit procedure need to define the responsibilities and requirements for planning and conducting audits, recording and reporting the audit results, follow up and implementation of corrective actions.
- The audit program is to be planned and executed. The planning of audits shall take into consideration the results of previous audits.
- The qualification of the auditors is one of the critical factors for successful audit program.
- The impartiality and conflict of interest for the auditors shall be considered when selecting the auditors.
Requirements for Internal Audit: ISO13485 Section 8.4
The audit records will include
1. Audit criteria,
2. Scope of the audit.
2. Interval,
3. Methods used for audit.
4. Conclusion of the audits.
5. Audit findings and criticality of the audit findings.
6. The status of the corrections and corrective actions.
7. Management review of the audit findings and status of the corrective actions.

Managing Internal Audit: (ISO13485 Section 8.4) in Qualcy eQMS

- You can do the audit planning and execution in the Qualcy eQMS
- The audit planning includes, scope, schedule and checklist to be used for the audit.
- You can create your own audit check list or use an inbuilt checklist. After the completion of the audits, the Qualcy eQMS provides option for review and approval of the audit reports.
- There is option for managing the audit non conformance and corrective actions for such nonconformance.
- There is option for reminders and auto notifications for the action plans.

Requirements for Nonconformance Management: ISO13485 Section 8.3
- You have to create documented procedures to manage the nonconforming product or process.
- The identification and segregation of nonconforming products are initial steps which must be done prior to the investigation.
- The investigation shall be done to confirm the validity of the non conformance, and scope of the nonconformance.
- If the products have been released to the customers, you will need to notify the customers and take actions to minimize the adverse impacts.
- You will need to conduct risk assessment to open CAPA to prevent reoccurrence of the nonconforming product.
- The records for the nonconforming product are to bemaintained (i.e., RF00XX: Out of Specification Report Form.
- When regulated products are involved, depending upon the classification and type of nonconformance, you will need to report to regulatory authorities (FDA/EMA) as appropriate.

Managing Nonconforming product/process: (ISO13485 Section 8.3) in Qualcy eQMS
- There is option for managing the corrective actions
- The identification of the nonconforming product or process, criticality of non-conformance, reference to the documents and previous records, review and approval of the conformance including disposition of the product.
- The details of the investigation done.
- There is option for managing the corrections and corrective actions for such non-conformance. The CAPA records can be linked to NC records.
- There is option for reminders and auto notifications for the action plans and pending approvals.
- There is option for metrics and trend analysis for the corrections and corrective actions.

Requirements for Corrective Actions: ISO13485 Section 8.5.2
- When nonconformance is observed, including product,process or customer complaints, you are required to take corrective actions to eliminate the cause of nonconformities to prevent the recurrence.
- The corrective actions shall be taken in a timely manner.
- The corrective actions will be proportionate to the risk of impact of the non conformance.
- You will need documented procedure for implementation of corrective action.
- You have to maintain the documented records for corrective actions taken and effectiveness of such actions.

Requirements for Corrective Actions: ISO13485 Section 8.5.2

The procedure for corrective actions shall include,
- Review of nonconformities including previous incidences and customer complaints, determining the root causes of nonconformities
- Implementation of the corrective actions to prevent recurrence of nonconformances (this may include updating specifications, drawings, batch records or standard
operating procedures as needed). - Verify that the corrective actions does not adversely impact the quality and performance of the products.
- Review of the effectiveness of the corrective action taken.
- Maintain the records for the investigation and the corrective action taken.

Requirements for Preventative Actions: ISO13485 Section 8.5.3
- You have to implement preventative actions to eliminate the potential non conformance before their occurrence. This can be done through review of the trends , feedbacks and others.
- You will need a documented procedure for handling of preventative action. This procedure can be combined with corrective action procedure.
- You have to maintain the documented records for preventive actions taken and effectiveness of such actions.

Requirements for Preventative Actions: ISO13485 Section 8.5.3

The PA(Preventative Action) procedure shall include:
- Determining potential non conformances and their causes.
- Evaluating the need for preventive actions to prevent occurrence of potential nonconformities.
- Planning, implementation and documentation of the preventative action taken. This may include updating specifications, batch records or standard operating
procedures as needed. - Verifying that the preventive actions do not adversely impact the quality and performance of the products.
- Review of the effectiveness of the preventive actions taken.
The records for the investigation and the preventive action taken shall be maintained.
Managing Corrective and Preventive Actions ( ISO13485 Section 8.5.2) in Qualcy eQMS
You can create CAPA(corrective and preventive actions) reports with reference links to other modules including NCs, Deviations, Equipment, Suppliers.
▪ You can assign tasks to other individuals for doing the investigations, implementation of corrective actions, effectiveness verification.
▪ Electronic review and approval of CAPA records.
▪ Easy access to metrics for overdue CAPAs, Tasks.
▪ Reminders and notifications for the open tasks and pending approvals.
▪ Easy access to workflow status visibility.
▪ Download and print CAPA reports, no separate CAPA form is required.